
Research Article | DOI: https://doi.org/10.31579/2640-1053/075
*Corresponding Author: Milan Radovich, PhD, IU Melvin and Bren Simon Comprehensive Cancer Center, IU School of Medicine, Michigan State, Indianapolis, IN 46202, USA.
Citation: Madison L. Conces, Bradley A. Hancock, R Atale, Jeffrey P. Solzak, Milan Radovich, et al. (2021) Whole-Genome Sequencing to Identify the Genetic Etiology of a Spontaneous Thymoma Mouse Model. Cancer Research and Cellular Therapeutics. 5(1); Doi: 10.31579/2640-1053/075
Copyright: © 2021, Milan Radovich, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 01 March 2021 | Accepted: 07 April 2021 | Published: 29 April 2021
Keywords: thymoma; mouse model; genomics; viral vector
Background: A mouse model for thymoma was previously created serendipitously by the random introduction of a transgene consisting of a mouse α-cardiac promoter, a constitutively active human transforming growth factor-β, and a simian virus 40 integration sequence into C3HeB/FeJ mice. Previous data demonstrated that the likely cause of thymomas in the thymoma mouse model was due to insertional mutagenesis by the transgene. At the time, fluorescence in situ hybridization was used to localize the transgene to the short arm of chromosome 2 (Chr2qF2-G region). In this exploratory study, we aimed to identify the exact insertion site of the transgene as this could provide clues to the genetic causation of thymomas in humans.
Materials and Methods: To identify the insertion site of the transgene, germline DNA from the thymoma mouse model was sequenced using low-pass, fragment-library, whole genome sequencing. Long-insert mate pair whole genome sequencing was employed to traverse the repetitive regions of the mouse’s genome and identify the integration site.
Results: The transgene was found to be integrated into a repetitive area of the mouse genome, specifically on Chr2qF1 within the intron of the FAM227B gene. Tandem integration of the transgene was observed with enumeration of an estimated 30 copies. Initial results suggested that a nearby gene, fibroblast growth factor 7 (Fgf7), could be affected by the gene insertion.
Conclusions: Whole genome sequencing of this thymoma mouse model identified the region of tandem integration of a transgene on Chr2qF1 that may have potential translational implications in helping to understand the genomic etiology of thymoma in humans.
The thymus is an organ located in the anterior-superior mediastinum that is critical to the development of the immune system after birth; subsequently, it atrophies or “involutes” in adolescence [1]. Current research to-date has not revealed a role of the thymus beyond childhood. However, thymic tissue can transform into a thymic tumor or “thymoma”. While a majority of early-stage thymomas have an indolent clinical course, a portion are able to metastasize leading to lethal outcomes. This is a rare malignancy, with approximately 500 cases in the United States each year, that affects both men and women equally [2]. Despite the relative rarity of this disease, it is the most common primary malignancy of the anterior-superior mediastinum [3].
Researchers at the Indiana University School of Medicine previously developed one of the only thymoma mouse model known to exist, named TIM-1 (thymoma insertional mutation) [4]. The original aim of the transgenic mouse model was to integrate a transgene consisting of an α-cardiac promoter, a constitutively active TGFβ gene, and simian virus (SV) 40 integration sequence for the study of hypertrophic cardiomyopathy (Figure 1). However, during the course of experimentation, several animals developed peripheral edema and labored breathing [4]. Postmortem examination revealed that the mice developed a neoplasm of epithelial origin that obstructed the great vessels resulting in the disease phenotype. These developments were uniformly fatal for all mice that inherited the transgene, 60-week mortality = 94%. Due to a lack of expression of the TGFβ transgene in the thymoma tissue, the observed phenotype was theorized to be caused by insertional mutagenesis. At that time, fluorescence in situ hybridization (FISH) was used and localized the transgene to the qF2-G region of chromosome 2 [4]. Further fine-mapping of the exact insertional site within the mouse genome and the identification of any potentially disrupted genes was not possible with the molecular biology techniques available at that time [4]. We hypothesized that if the insertion site of the transgene could be located within the genome of the TIM-1 mouse model, genes affected by the transgene insertion may provide valuable information regarding a potential genetic causation of thymomas in humans. Herein, using whole genome next-generation sequencing, we sought to understand the genetic etiology of thymoma in the TIM-1 mouse model.

TIM-1 DNA
TIM-1 germline DNA was obtained from the blood of three heterozygous C3HeB/FeJ mice [4]. Homozygous inheritance of the transgene is embryonically lethal; thus, blood from these mice was not used. Before drawing blood, the heterozygosity of the mouse was verified using a conventional PCR assay from an ear punch of each mouse to detect the presence of human TGFβ from mouse DNA samples. DNA was extracted from blood using the QIAmp DNA Mini Kit (Qiagen). RNase was used to help increase the purity of the DNA extracted.
TIM-1 Mouse Model
This study was carried out in strict accordance with the recommendations in the "Guide for the Care and Use of Laboratory Animals" of the National Institutes of Health. This protocol was approved by the Institutional Animal Care and Use Committee of Indiana University (IACUC), Protocol #10948. Standard protocols for housing, feeding, and environmental enrichment for all mice on this project were followed based on IACUC guidelines [5]. For genotyping purposes, ear punches were taken and anesthetic in the form of 1-5% isoflurane inhalation to effect with waste gas scavenging used. Visual monitoring during the procedure was performed and documentation of monitoring noted in the surgery log.
Low-pass fragment-library whole genome sequencing (WGS) and analysis
For library preparation, fragmentation was performed using the Ion Xpress plus Fragment Library Kit (Life Technologies) per manufacturer’s instructions. The incubation time for the fragmentation step was 5 minutes. Size selection was performed for DNA lengths between 270-370bp, per the manufacturer’s instructions. The combined library was diluted to a 10 pM concentration for loading into the emulsion PCR. The template preparation was performed using the Ion PI Template OT2 200 Kit v3 (Life Technologies) and was then quantified using the IonSphere Quality Control Kit (Life Technologies). For sequencing, the Ion PI Sequencing 200 Kit v3 Kit (Life Technologies) was used with the Ion Proton Sequencer and an Ion PI Chip Kit v2 (Life Technologies). The raw data collected from sequencing was analyzed using Torrent Suite 4.0.2 and provided as unmapped reads. Sequencing data was uploaded to the IU Quarry Supercomputer for analysis. The transgene reference sequence has been published previously and consisted of a mouse α-cardiac MHC promoter, human TGFβ cDNA, and a simian virus (SV) polyA sequence [4]. The Blast-like alignment tool (BLAT) [6] and basic local alignment search tool (BLAST) [7] were used to identify split reads that mapped to both the transgene sequence and the mouse reference genome.
Long-insert mate-pair WGS and analysis
Mate-pair sequencing libraries were generated with the Nextera Mate Pair Library Prep Kit (FC-132-1001, Illumina). The libraries were sequenced, utilizing 2x125bp reads on one lane of a HiSeq 2500. Bioinformatic pre-processing of the sequencing data included ab initio removal of duplicates, using an in-house tool at the New York Genome Center; low quality and adapter trimming, junction adapter clipping, using Cutadapt v1.8.1 [8]; and phiX spike-in removal, using GEM mapper pre-release 3 [9]. Putative integration sites were determined by extracting read pairs with at least one of the mates mapping either to hTGF-β or to the mMYH6 promoter and subsequently re-mapping them using BWA-MEM against the mouse reference genome (mm10) [10].
TIM-1 mice harboring the transgene develop pronounced hyperplasia of the thymus (Figure 2). The enlarging thymoma causes obstruction of the great vessels, leading to severe peripheral edema with subsequent death attributed to rupture of the vessels. Liver congestion is observed in these mice that may be secondary to the great vessel obstruction. Previous analyses have shown that female mice have a considerably inferior overall survival compared to male mice for reasons that are not well understood.

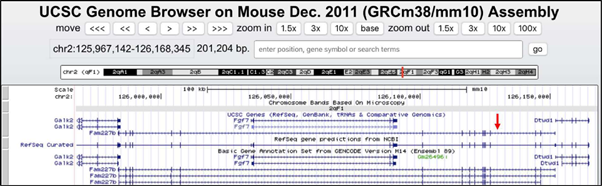
Low-pass, fragment WGS produced 6.75x coverage of the TIM-1 genome. Reads were first mapped to the transgene sequence. Those reads were then isolated and re-mapped to the mouse reference genome to identify split reads that may lend clues to genomic localization. Confirmation of the presence of the transgene was observed as evidenced by reads mapping to the mutated human TGF-β portion of the transgene (data not shown). Reads were also identified supporting tandem integration by identifying split reads that map to both the end and the start of an adjoining transgene (data not shown). Unfortunately, no reads were able to be identified that mapped to both the transgene and uniquely to the mouse chromosome 2F-G region. Many of the split reads mapped to highly repetitive areas of the genome. An inspection of the chromosome 2F-G region using UCSC genome browser demonstrated a significant presence of repetitive regions. We hypothesized that the transgene may have integrated into a repetitive region and would require an alternate method that could traverse long stretches of repetitive elements. Long-insert mate pair sequencing enables paired-end sequencing with larger (several kb) inserts that can traverse longer stretches of the genome compared to fragment-based sequencing (11). Whole-genome long-insert mate pair sequencing was performed on TIM-1 germline DNA to an average coverage of 30X. The modal insert size of the mate-pair libraries was approximately 5 kb. With the use of long-insert mate base pair sequencing, the insertion site was identified to be specifically located in Chr2qF1 (Figures 3A, 3B). The approximate position was noted to be chr2:126,129,000 (mm10 reference genome). This region is located within an intron of the gene Fam227b. A review of mapped reads with IGV demonstrated >150X coverage of the MHC promoter, signifying the likely tandem integration of the transgene (Figure 4). We estimate that approximately thirty tandem copies of the transgene were inserted into the TIM-1 genome. With the use of the UCSC genome browser, a gene called Fgf7 (fibroblast growth factor 7) that plays a well-known role in thymus development was noted to be near the insertion site of the transgene (Figure 5). Pending submission to publicly available repository.



Thymoma is a rare malignancy with few experimental models available to study the biology of the disease. The TIM-1 mouse represents one of the only in vivo models of spontaneous thymoma to date [12-14]. While serendipitous creation of this mouse by random insertional mutagenesis of a transgene created a much-needed preclinical model, the exact genetic disruption could not previously be elucidated due to the lack of genomic technologies that could localize the insertion [4]. Although the transgene contains the cDNA sequence of a constitutively active TGF-β [by a Cys33Ser mutation], this is not thought to be the driver of thymoma development, as TGF-β RNA was not detected in the thymoma tissue[4]. Through the use of long-insert mate pair whole-genome sequencing, we were able to locate the insertion site of the transgene down to the gene-level. Our analysis revealed it to be located on chromosome 2 of the murine genome within an intron of the FAM227B gene. This insertion is likely present as a long chain of tandem repeats of the transgenes, with an estimate of 30 copies.
The FAM227B gene has an unknown function. However, FAM227B in both the human and murine genomes is oriented in an antisense fashion to another gene known to potently regulate thymic development: Fgf7 [8]. Although the insertion does not overlap Fgf7 on the antisense strand, it is present ~40kb downstream in the 3’ direction (Figure 5). Whereas the physiology of FAM227B is yet to be characterized, we can speculate about its potential relationship to genes known to be important in thymic function and begin to deduce why the transgene insertion had such deleterious effects. A differential expression analysis of Fgf7 was attempted but was ultimately not feasible due to the small size and histological composition of the tissues.
Prior studies have shown that Fgf signaling is important in the regulation of the developing thymus and for thymopoiesis. Key players of Fgf signaling involved in thymic development include Fgf7, Fgf10, and Fgfr2IIIb [15]. Fgf7 and Fgf10 belong to the group of ligands known as fibroblast growth factors [Fgf] and are expressed in the fetal thymus. A receptor for these ligands, Fgf receptor-2 IIIb [Fgfr2IIIb], is expressed on thymic epithelial cells and in the developing thymus as well [15, 16]. One study suggests that Fgfr2IIIb may have a greater role in the development of the thymus than how much Fgf7 and Fgf10 are available to bind [17]. In addition, bone morphogenic protein [BMP] functions upstream of both Fgf7 and Fgf10, to regulate T-cell development and enhance Fgfr2IIIb expression. This may increase the sensitivity of thymic epithelial cells to Fgf7 and Fgf10 stimulation in the development of the thymus [18]. Finally, the administration of Fgf7 may also have the ability to improve weakened immune systems, such as in elderly individuals, linking it further to thymoma function and development[19-21]. Ink4a, an important regulator in thymopoiesis and a tumor suppressor gene, impairs the proliferation of early T cell progenitors when overexpressed [19, 20]. Fgf7 has been found to downregulate Ink4a, resulting in the rejuvenation of early T cell progenitors [21]. As a result of our study, it is reasonable to further postulate that dysregulation in the Fgf7/FAM227B region in addition to aberrant Fgf signaling likely has significant implications for thymic neoplastic development and may also be targetable using clinically available inhibitors of Fgf signaling.
While this study suggests where the insertion site of transgene may be, a knockout mouse was not created to attempt to reproduce these findings. Developing a mouse model with an inactivated FAM227B gene would be a future project that could be undertaken to further evaluate whether the insertion of this transgene did cause these mice to develop thymomas.
Herein, we genetically define the TIM-1 mouse model of spontaneous thymoma, providing a useful resource to the thymic malignancy research community. We postulate that the insertion of the transgene may be driving the overexpression of Fgf7 leading to thymoma. Ultimately, a deeper understanding of the genetic drivers of thymoma will facilitate development of biologically-informed therapies for this rare disease
Dr. Loehrer is supported by NCI grants P30 CA082709 and U54CA190151.
Conflicts of Interest: The authors have no conflicts of interest to declare.
Financial Disclosure: We would like to acknowledge the generous support for these studies from the William P. Loehrer fund at the Indiana University Melvin and Bren Simon Cancer Center.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
